
NGS data analysis.
Silencing Genomes. Python. Bioinformatics tools. Journals. PHP. UML. Genostar Tools. Genostar's bioinformatics data analysis platform enables researchers to explore, interpret, compare and analyze life sciences data at the genomic, proteic and metabolic levels.
Our goal is to help researchers find relevant links in the complex network of genomes, proteomes and pathways involved in cellular processes. Next Generation Sequencing and high throughput expression analysis are rapidly growing areas. Genostar's software and data are designed to analyze and manage a wide range of experimental data, from NGS-generated sequences to gene expression results.
In addition to facilitating extensive comparative analyses, our MicroB database also makes it possible for researchers and organizations to save and centralize their analyzed genomes. Sequencher Features. Gene Codes has long been an innovator, investing in the R&D to develop powerful features for your DNA sequence analysis.
Gene Codes developed the Assemble to Reference Sequence strategy that is widely used to speed up assembly and assign base-numbering systems and features to new data. The variance table was developed in the mid 1990's and became a key element first for forensic sequencing of mtDNA, and then for virtually all of our collaborators. Working with core labs that use structured naming conventions to track data for individual clients, we developed the Assemble by Name strategy that has become such a powerful and popular tool for combining multiple sequencing projects into a single analysis run. We have continued our strategy of adding functionality to Sequencher that focuses on labs doing DNA sequencing. Look through some of the topics at the left and explore some of the features, both common and specialized, that are in Sequencher today.
A Post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. The Elements of Bioinformatics. ABACAS: algorithm-based automatic contiguation of assembled sequences. PAGIT - Post Assembly Genome Improvement Toolkit - Wellcome Trust Sanger Institute. Tools to generate automatically high quality sequence by ordering contigs, closing gaps, correcting sequence errors and transferring annotation.

An Explanation of Velvet Parameter exp_cov - Homologus. Homolog.us blog is written by professional janitors dedicated to clean up US science.
During lunch breaks and other time off from the job, we discuss bioinformatics. The name 'homolog.us' is not a spelling mistake, but is derived by taking Arabic translation of the 'O' in the original word. Please follow us on twitter – @homolog_us. Appropriate choice of the ‘exp_cov’ (expected coverage) parameter in Velvet is very important to get an assembly right. In the following figure, we show data from a calculation on a set of reads taken from a 3Kb region of a genome, and reassembling them with varying exp_cov parameters. As you can see from the above figure, when exp_cov is low, Velvet has difficulty reassembling the original sequence.
Why does the assembly change so dramatically with exp_cov parameter? I) Unique region – The sequence, where the read comes from in the genome, is unique region. Breakdancer - Bioinformatics Team (BioITeam) at the University of Texas. Skip to end of metadataGo to start of metadata.
PyMol Tutorial. [return to KB Wong's home page] A tutorial on using PyMOL to generate publication quality figures. Through this tutorial, you will be able to generate the following figures: You can download the files for the tutorials here . Crystallography & Biocomputing Group. First and basic command is "ray".
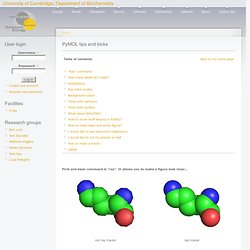
It allows you to make a figure look nicer... Online Lectures on Bioinformatics. Next Generation Genomics - Velvet Assembly Workshop. Next Generation Genomics.

Locations. Rosalind is a platform for learning bioinformatics and programming through problem solving.
Take a tour to get the hang of how Rosalind works. If you don't know anything about programming, you can start at the Python Village.
De-novo sequencing. R programming. Mathematics. Algorithms. The Bioinformatics Adventure. Software packages for next gen sequence analysis [Archive] 28 Dec 2009: This thread has been closed.
![Software packages for next gen sequence analysis [Archive]](http://cdn.pearltrees.com/s/pic/th/software-packages-seqanswers-50225738)
Please see our wiki software portal ( for information about each of these packages. A reasonably thorough table of next-gen-seq software available in the commercial and public domain Integrated solutions * CLCbio Genomics Workbench ( - de novo and reference assembly of Sanger, Roche FLX, Illumina, Helicos, and SOLiD data. Commercial next-gen-seq software that extends the CLCbio Main Workbench software. Includes SNP detection, CHiP-seq, browser and other features. Next Generation Sequencing and Data Analysis. CORTEX website. 13th November 2013: release v1.0.5.21.

Get it here. Minor release with updates to the 1000 Genomes Phase3 pipeline. Emacs. Releases | Supported Platforms | Obtaining Emacs | Documentation | Support | Further information GNU Emacs is an extensible, customizable text editor—and more. At its core is an interpreter for Emacs Lisp, a dialect of the Lisp programming language with extensions to support text editing. The features of GNU Emacs include: Content-sensitive editing modes, including syntax coloring, for a variety of file types including plain text, source code, and HTML.Complete built-in documentation, including a tutorial for new users.Full Unicode support for nearly all human languages and their scripts.Highly customizable, using Emacs Lisp code or a graphical interface.A large number of extensions that add other functionality, including a project planner, mail and news reader, debugger interface, calendar, and more.
Many of these extensions are distributed with GNU Emacs; others are available separately. Releases Emacs 24 has a wide variety of new features, including: Supported Platforms. Galaxy. Snptest. SNPTEST is a program for the analysis of single SNP association in genome-wide studies. The tests implemented include. IMPUTE. [2] B. N. BEAGLE genetic analysis software. Program: BEAGLE Version: 3.3.2 Copyright (c) 2007-2013 Brian L. Browning Email: browning@uw.eduThis page was last updated on 11 Oct 2013 Contents Introduction. PLINK: Whole genome data analysis toolset.
Bioinformatics Applications. This knowledge book describes bioinformatic applications applicable to the IBEST core facilities. Description "The Environment Modules package provides for the dynamic modification of a user's environment via modulefiles. Each modulefile contains the information needed to configure the shell for an application. Once the Modules package is initialized, the environment can be modified on a per-module basis using the module command which interprets modulefiles. Typically modulefiles instruct the module command to alter or set shell environment variables such as PATH, MANPATH, etc. modulefiles may be shared by many users on a system and users may have their own collection to supplement or replace the shared modulefiles.
GATK Support Forum. GATK Main Page. Next Genetics. SEQanswers Home.
Bioinformatics Answers.