
„Nazywam się Paasche, byłem oficerem marynarki, a teraz jestem rewolucjonistą” Berlin, 9 listopada 1918. Locations. Rosalind is a platform for learning bioinformatics and programming through problem solving.
Take a tour to get the hang of how Rosalind works. If you don't know anything about programming, you can start at the Python Village. For a collection of exercises to accompany Bioinformatics Algorithms book, go to the Textbook Track. Otherwise you can try to storm the Bioinformatics Stronghold right now. If you are completely new to programming, try these initial problems to learn a few basics about the Python programming language. Bioinformatics Stronghold Discover the algorithms underlying a variety of bioinformatics topics: computational mass spectrometry, alignment, dynamic programming, genome assembly, genome rearrangements, phylogeny, probability, string algorithms and others. Ready-to-use software tools abound for bioinformatics analysis. Bioinformatics Textbook Track. Omics - Omics.org. National Resource for Network Biology. The genetic code is nearly optimal for allowing additional information within protein-coding sequences.
+ Author Affiliations Abstract DNA sequences that code for proteins need to convey, in addition to the protein-coding information, several different signals at the same time.

These “parallel codes” include binding sequences for regulatory and structural proteins, signals for splicing, and RNA secondary structure. Here, we show that the universal genetic code can efficiently carry arbitrary parallel codes much better than the vast majority of other possible genetic codes. This property is related to the identity of the stop codons. Footnotes ↵3 Corresponding author. ↵3 E-mail uri.alon@weizmann.ac.il; fax 972-8-934125.
GeneticCode.pdf (application/pdf Object) Stephen Jay Gould. Stephen Jay Gould (/ɡuːld/; September 10, 1941 – May 20, 2002) was an American paleontologist, evolutionary biologist and historian of science.
He was also one of the most influential and widely read writers of popular science of his generation.[1] Gould spent most of his career teaching at Harvard University and working at the American Museum of Natural History in New York. In the later years of his life, Gould also taught biology and evolution at New York University. Gould's most significant contribution to evolutionary biology was the theory of punctuated equilibrium, which he developed with Niles Eldredge in 1972.[2] The theory proposes that most evolution is marked by long periods of evolutionary stability, which is punctuated by rare instances of branching evolution. The theory was contrasted against phyletic gradualism, the popular idea that evolutionary change is marked by a pattern of smooth and continuous change in the fossil record. Biography[edit] Kodon. Tabela kodonów Fakt, iż kodonów jest więcej niż aminokwasów nazywamy degeneracją kodu genetycznego (kilka kodonów może dyktować ten sam aminokwas).
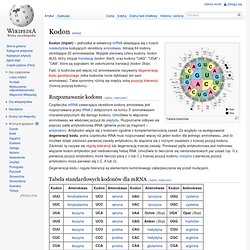
Takie synonimy różnią się między sobą pozycją tolerancji (trzecią pozycją kodonu). Rozpoznawanie kodonu[edytuj | edytuj kod] Cząsteczka mRNA zawierająca określone kodony aminokwas jest rozpoznawana przez tRNA z dołączonym na końcu 3' aminokwasem charakterystycznym dla danego kodonu. Umożliwia to włączenie aminokwasu we właściwej pozycji do peptydu. Degeneracja kodu i reguła tolerancji są elementami komórkowego zabezpieczania się przed mutacjami. Tabela standardowych kodonów dla mRNA[edytuj | edytuj kod] 1 UGA – w mitochondriach i u niektórych jednokomórkowych protistów koduje tryptofan; 2 AUG – u Prokaryota występuje formylometionina; 3 GUG – alternatywny kodon Start niektórych Prokaryota. Sequence alignment. A sequence alignment, produced by ClustalW, of two humanzinc finger proteins, identified on the left by GenBank accession number.
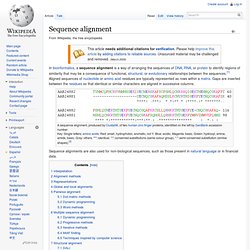
Key: Single letters: amino acids. Red: small, hydrophobic, aromatic, not Y. Blue: acidic. Magenta: basic. Green: hydroxyl, amine, amide, basic. Sequence alignments are also used for non-biological sequences, such as those present in natural language or in financial data. Interpretation[edit] Alignment methods[edit] Very short or very similar sequences can be aligned by hand. Representations[edit] Sequence alignments can be stored in a wide variety of text-based file formats, many of which were originally developed in conjunction with a specific alignment program or implementation. Global and local alignments[edit] Illustration of global and local alignments demonstrating the 'gappy' quality of global alignments that can occur if sequences are insufficiently similar Pairwise alignment[edit] Dot-matrix methods[edit]
European Bioinformatics Institute. FASTA format. Format FASTA jest formatem zapisu sekwencji kwasów nukleinowych oraz białek używanym w bioinformatyce.
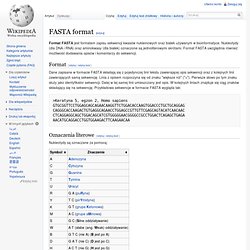
Nukleotydy (dla DNA i RNA) oraz aminokwasy (dla białek) oznaczone są jednoliterowymi skrótami. Format FASTA uwzględnia również możliwość dodawania opisów i komentarzy do sekwencji. Format[edytuj | edytuj kod] Dane zapisane w formacie FASTA składają się z pojedynczej linii tekstu zawierającej opis sekwencji oraz z kolejnych linii zawierających samą sekwencję. Linia z opisem rozpoczyna się od znaku "większe niż" (">").
>Keratyna 5, egzon 2, Homo sapiens GTGCGGTTCCTGGAGCAGCAGAACAAGGTTCTGGACACCAAGTGGACCCTGCTGCAGGAG CAGGGCACCAAGACTGTGAGGCAGAACCTGGAGCCGTTGTTCGAGCAGTACATCAACAAC CTCAGGAGGCAGCTGGACAGCATCGTGGGGGAACGGGGCCGCCTGGACTCAGAGCTGAGA AACATGCAGGACCTGGTGGAAGACTTCAAGAACAA Oznaczenia literowe[edytuj | edytuj kod] Nukleotydy są oznaczane za pomocą: Aminokwasy są określane za pomocą: