
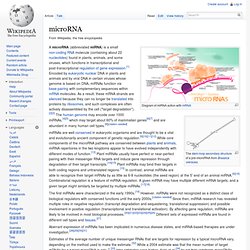
Gene-Quantification.info - The reference in qPCR - Academic & Industrial Information Platform. MirTools: a web server for microRNA profilling and discovery based on high-throughput sequencing. MicroRNA. Diagram of miRNA action with mRNA miRNAs are well conserved in eukaryotic organisms and are thought to be a vital and evolutionarily ancient component of genetic regulation.[9][10][11][12] While core components of the microRNA pathway are conserved between plants and animals, miRNA repertoires in the two kingdoms appear to have evolved independently with different modes of function.[13] Plant miRNAs usually have perfect or near-perfect pairing with their messenger RNA targets and induce gene repression through degradation of their target transcripts.[14][15] Plant miRNAs may bind their targets in both coding regions and untranslated regions.[15] In contrast, animal miRNAs are able to recognize their target mRNAs by as little as 6-8 nucleotides (the seed region) at the 5' end of an animal miRNA.[6][16] Combinatorial regulation is a feature of miRNA regulation.
History[edit] MiRBase. MiRDeep2. Discovering known and novel miRNAs from deep sequencing data Developed by Sebastian Mackowiak, Marc Friedländer and Nikolaus RajewskySystems Biology group at the Max Delbrück Center, Berlin-BuchApril 20th, 2011 miRDeep2 is a completely overhauled tool which discovers microRNA genes by analyzing sequenced RNAs.
The tool reports known and hundreds of novel microRNAs with high accuracy in seven species representing the major animal clades. The low consumption of time and memory combined with user-friendly interactive graphic output makes miRDeep2 accessible for straightforward application in current reasearch. Download: miRDeep2.0.0.5 contains the core algorithm script and all auxiliary scripts. The package is freely available for non-commercial purposes. BioinfoUGR. sRNAbench is a free web-server tool and standalone application for processing small-RNA data obtained from next generation sequencing platforms such as Illumina or SOLiD.
The sRNAbench tool is the replacement for miRanalyzer. The full manual is availabe here. MiRanalyzer. Rfam 11.0: 10 years of RNA families. + Author Affiliations ↵*To whom correspondence should be addressed.

MiRWalk - The Database on Predicted and Published MicroRNAs. How to cite miRWalk database?

Dweep, H., Sticht, C., Pandey, P., Gretz, N., miRWalk - database: prediction of possible miRNA binding sites by "walking" the genes of 3 genomes, Journal of Biomedical Informatics, 44: 839-7, 2011. A software for the analysis of microRNA Deep Sequencing data. Introduction Deep Sequencing technologies (Next Generation Sequencing) are increasingly used to obtain a more accurate depiction of miRNA expression profiles in living cells.

This type of analysis is a key step towards improving our understanding of the complexity and mode of miRNA regulation. miRNAkey is a software package designed to be used as a base-station for the analysis of miRNA deep sequencing data. Our package implements common steps taken in the analysis of such data, as well as adds unique features, such as data statistics and multiple mapping levels, generating a novel platform for the analysis of miRNA expression.
Through the use of a simple graphical interface, the user can determine the analysis steps. The tabular and graphical output contains detailed reports on the sequence reads and provides an accurate picture of the differentially expressed miRNAs in paired samples. Run miRNAkey Make sure all the requirements are properly installed. IsoMIRex. MiRPara Online. Abstract: BACKGROUND:MicroRNAs are a family of ~22nt small RNAs that can regulate gene expression at the post-transcriptional level.

Identification of these molecules and their targets can aid understanding of regulatory processes. Recently, high throughput sequencing (HTS) has become a common identification method but there are two major limitations associated with the technique. Firstly, the method has low efficiency, with typically less than 1 in 10,000 sequences representing miRNA reads and secondly the method preferentially targets highly expressed miRNAs. If sequences are available, computational methods can provide a screening step to investigate the value of an HTS study and aid interpretation of results. RNAdb 2.0 - A database of mammalian noncoding RNAs. In June 2012, the RNAdb 2.0 database was officially retired.

RNAdb 2.0 was published in Nucleic Acids Research in 2007 with the following citation: Pang, KC, Stephen, S, Dinger, ME, Engstrom, PG, Lenhard, B, Mattick, JS (2007). RNAdb 2.0--an expanded database of mammalian non-coding RNAs. Nucleic Acids Res 35: D178-182. This website serves to archive the final snapshot of the RNAdb 2.0 datasets for legacy purposes. A suite of tools for analysing micro RNA and other small RNA data from High-Throughput Sequencing devices.
Arraystar. The high throughput sequencing technique provides high sensitivity and specificity to analyze the abundance of microRNA sequence in a sample as well as to discover novel microRNA species.

Arraystar offers Integrated microRNA Sequencing Service from sequencing library preparation to comprehensive data anlaysis. Our data analysis process generally consists of the following steps, raw data processing, usable reads filtering (including 3' adapter trimming and normalization), and bioinformatics analysis( fig. 1). MirTools: a web server for microRNA profilling and discovery based on high-throughput sequencing. A step-by-step usage guideline for mirTools Users can navigate the platform in two modes: Based on individual sample Under this mode, only one sample is allowed for easy small RNA annotation.
Users need to wait for the result by Email. When the job is finished, an Email will be sent to you with kind notification. Step 1: users are requested to enter their email address, through which automatically generated job ID number can be delivered to customer for further result retrieve. MAGI - DBMI @ UCSD. MiRNA annotation tools comparison. MiRNA annotation tools comparison In summary: I will show which is the best miRNA mapping tool. I used 4 options for this benchmarking: I think that these are the most used. They were clearly developed for other purposes, but as well, they generate the input of many miRNA pipelines. I just wanted to know how well my tool was doing. The first aim to develop miraligner was to get annotated additions of nucleotides at the end of miRNA sequences, something that is very common in mirna biogenesis: isomirs and often they are missed by short read and fast mappers. I have a repository for this kind of things, so anybody can reproduce my results, and check if I did something wrong, or comment on it.
GEN220_sRNA_seq.pdf. Comments.