
Tag: Sequence Alignment. Tag: Bioinformatics. Genebase. M i c r o b e s . n l. Sequence Alignment Techniques. Sequence Alignment Techniques are very useful in the field of Bioinformatics.
Bioinformatics is the application of computer technology in the field of Biology. Within the nucleus of our cells, a very fine thread like structure is present known as chromosome. The chief component of chromosome is the DNA( Deoxyribonucleic acid). Without getting into much detail, just know this that the DNA can be represented as a string of letters. Example- ACCTGATCGATCAGTGACGAT, such strings are known as DNA sequences. Let us align 2 DNA sequences. Suppose the sequences to be compared are much more complex, say Seq1- ABCNYRQCLCRPM( Query Sequence )Seq2- AYCYNRCKCRBP( Subject Sequence ) In such cases, we use algorithms; 2 main algorithms used are Needleman-Wunsch algorithm and Smith-Waterman algorithm. =>Initiation of the matrix Before we initiate the matrix we have to assign the match value, mismatch value and gap value. Background on Nucleic Acid Dot Plots. Dot plots provide a simple, yet extremely powerful means of nucleic acid sequence analysis.

Most commonly they are examined to identify one of two situations: Regions of similarity within a single molecule (i.e. repeats) or between different molecules. Sequences that have the potential for forming secondary structure by intramolecular base-pairing The principle used to generate dot plots is straightforward. A matrix comparison of two sequences (or one with itself) is prepared by "sliding" a window of user-defined size along both sequences. In the following example, a mismatch limit of 2 means that up to 2 of 5 bases may mismatch and the 5 base window will still be classified as a match; when mismatch limit is set to 0, all 5 bases must match perfectly in order for the windows to match. List of sequence alignment software. JAligner: Java implementation of the Smith-Waterman algorithm for biological sequence alignment. FASTA. FASTA is a DNA and protein sequence alignment software package first described (as FASTP) by David J.
Lipman and William R. Pearson in 1985.[1] Its legacy is the FASTA format which is now ubiquitous in bioinformatics. Smith–Waterman algorithm. The Smith–Waterman algorithm performs local sequence alignment; that is, for determining similar regions between two strings or nucleotide or protein sequences.
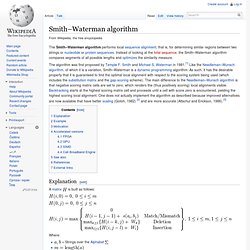
Instead of looking at the total sequence, the Smith–Waterman algorithm compares segments of all possible lengths and optimizes the similarity measure. The algorithm was first proposed by Temple F. Smith and Michael S. Waterman in 1981.[1] Like the Needleman–Wunsch algorithm, of which it is a variation, Smith–Waterman is a dynamic programming algorithm. As such, it has the desirable property that it is guaranteed to find the optimal local alignment with respect to the scoring system being used (which includes the substitution matrix and the gap-scoring scheme). Explanation[edit] is built as follows: Where: Hirschberg's algorithm. Algorithm information[edit]
Needleman–Wunsch algorithm. The Needleman–Wunsch algorithm is an algorithm used in bioinformatics to align protein or nucleotide sequences.
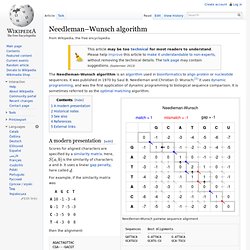
It was published in 1970 by Saul B. Needleman and Christian D. Wunsch;[1] it uses dynamic programming, and was the first application of dynamic programming to biological sequence comparison. It is sometimes referred to as the optimal matching algorithm. Needleman-Wunsch pairwise sequence alignment Sequences Best Alignments --------- ---------------------- GATTACA G-ATTACA G-ATTACA GCATGCU GCATG-CU GCA-TGCU A modern presentation[edit] Scores for aligned characters are specified by a similarity matrix. Sequence alignment. A sequence alignment, produced by ClustalW, of two humanzinc finger proteins, identified on the left by GenBank accession number.
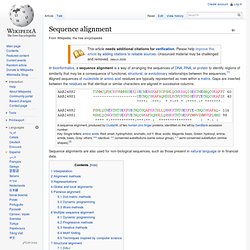
Key: Single letters: amino acids. Red: small, hydrophobic, aromatic, not Y. Blue: acidic. BLAST. In bioinformatics, BLAST for Basic Local Alignment Search Tool is an algorithm for comparing primary biological sequence information, such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences.
A BLAST search enables a researcher to compare a query sequence with a library or database of sequences, and identify library sequences that resemble the query sequence above a certain threshold. Different types of BLASTs are available according to the query sequences. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.
The BLAST program was designed by Stephen Altschul, Warren Gish, Webb Miller, Eugene Myers, and David J. Lipman at the NIH and was published in the Journal of Molecular Biology in 1990.[1] Input[edit]