
Bundesverband für Neurofibromatose. Neurofibromatose Typ 2. NF-2 Locus Die Neurofibromatose Typ II (NF II), auch als zentrale Neurofibromatose bezeichnet, ist eine erbliche Tumorerkrankung.
Ihr Hauptmerkmal ist das Vorkommen von gutartigen Hirntumoren, die sich symmetrisch im Bereich beider Hör- und Gleichgewichtsnerven entwickeln. Die meisten Patienten mit dieser Erkrankung leiden auch an Veränderungen der Augen. Ursache der NF II sind Mutationen eines Gens, das vermutlich Einfluss nimmt auf Form und Wanderungsverhalten bestimmter Zelltypen.
Da die NF II genetisch bedingt ist, ist eine Heilung nicht möglich. Neurofibromatosis type II. Neurokutane Erkrankung. Die Neurokutanen Erkrankungen (Synonym: Phakomatosen, Neuroectodermale Erkrankungen) sind charakterisiert durch Manifestationen an den beiden Organen Haut und Nervensystem.
Die Klassifikation dieser Erkrankungsgruppe variiert je nach Lehrbuchautor. Varianten der Klassifikation[Bearbeiten] Das Lehrbuch der Neurologie von Mumenthaler (1979) definiert die Phakomatosen als Fehlbildungen an zentralem Nervensystem und Haut und zählt hierzu die Neurofibromatose Recklinghausen (NF), die Tuberöse Sklerose Bourneville-Pringle (TS), die Encephalo-Faciale Angiomatose Sturge-Weber (EFA) und die Retino-Zerebelläre Angiomatose Hippel-Lindau (RZA). Das Lehrbuch der Neurologie von Delank (1994) definiert die neurokutanen Erkrankungen aufgrund von histologischen und embryologischen Überlegungen. Dort heißt es, die Phakomatosen seien dysplastisch-blastomatöse Entwicklungsstörungen, die ektodermale Strukturen betreffen und somit als neurokutane Erkrankungen auftreten. Neurofibromatose Typ 1. Typische Veränderungen an der Haut sind mehrere Café-au-lait-Flecken sowie Neurofibrome.
Im zentralen Nervensystem (ZNS) treten gehäuft Tumore verschiedener Lokalisation auf. Patienten können minderbegabt sein und an epileptischen Anfällen leiden. Des Weiteren sind regelmäßig Augen und Knochen mitbetroffen. Eine Neurofibromatose wird durch eine Veränderung in einem Gen hervorgerufen, welches normalerweise hemmend auf die Zellteilung Einfluss nimmt.
Es kommt daher zu überschießender Gewebsvermehrung und damit zu den typischen Veränderungen. Neurofibromatosis type I. Patient with multiple small cutaneous neurofibromas and a 'café au lait spot' (bottom of photo, to the right of centre).
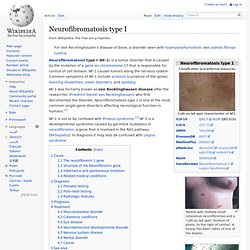
A biopsy has been taken of one of the lesions. Back of an elderly woman with NF-1 Neurofibromatosis type I (NF-1) is a tumor disorder that is caused by the mutation of a gene on chromosome 17 that is responsible for control of cell division. NF-1 causes tumors along the nervous system. Common symptoms of NF-1 include scoliosis (curvature of the spine), learning disabilities, vision disorders, and epilepsy. Neurofibroma. Subtypes of Neurofibroma[edit] Dermal Neurofibroma[edit] Anatomy[edit] Dermal neurofibromas (sometimes referred to as cutaneous neurofibromas) originate in nerves in the skin.
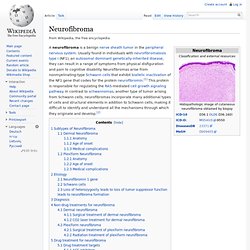
Three kinds are distinguished:[3] Discrete cutaneous neurofibromas: Sessile or pedunculated masses on the skin, which are fleshy and non-tender, and can vary in size.Discrete subcutaneous neurofibromas: Lie below and look like bumps on the skin, which can sometimes be tender.Deep nodular neurofibromas: Involving tissues and organs underneath the dermis, but otherwise resembling cutaneous and subcutaneous neurofibromas. Neurofibrom. Neurofibrom der Haut, mikroskopisches Bild Neurofibrome der Haut, makroskopisches Bild Neurofibrome sind Nerventumore, die innerhalb des Nervs gut abgrenzbare oder außerhalb des Nervs diffuse Strukturen ausbilden.
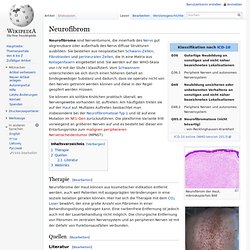
Sie bestehen aus neoplastischen Schwann-Zellen, Fibroblasten und perineuralen Zellen, die in eine Matrix aus Kollagenfasern eingebettet sind. Sie werden auf der WHO-Skala von I-IV mit der Stufe I klassifiziert. Vom Schwannom unterscheiden sie sich durch einen höheren Gehalt an bindegewebiger Substanz und dadurch, dass sie operativ nicht von den Nerven getrennt werden können und diese in der Regel geopfert werden müssen.
Sie können als solitäre Knötchen praktisch überall, wo Nervengewebe vorhanden ist, auftreten. Therapie[Bearbeiten] Neurofibrome der Haut können aus kosmetischer Indikation entfernt werden, auch weil Patienten mit ausgeprägten Veränderungen in eine soziale Isolation geraten können. Quellen[Bearbeiten] Literatur[Bearbeiten] Drappier et al. Neurofibromatose. Neurofibromatose Typ 1 (NF1) (Morbus Recklinghausen, periphere Neurofibromatose)Neurofibromatose Typ 2 (NF2) (zentrale Neurofibromatose)Hochspringen ↑ Werner Buselmaier, Gholamali Tariversian: Humangenetik. 4., neu bearbeitete Auflage.
Springer, Berlin 2006, ISBN 3-540-32677-4, S. 183, hier online. Neurofibromatosis. Neurofibromatosis (NF) refers to a number of inherited conditions that are clinically and genetically distinct and carry a high risk of tumor formation, particularly in the brain.[1] Neurofibromatosis is an autosomal dominant disorder, which means only one copy of the affected gene is needed for the disorder to develop.
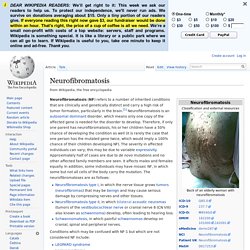
Therefore, if only one parent has neurofibromatosis, his or her children have a 50% chance of developing the condition as well (it is rarely the case that one person has the mutated gene twice, which would imply a 100% chance of their children developing NF). The severity in affected individuals can vary; this may be due to variable expressivity. Approximately half of cases are due to de novo mutations and no other affected family members are seen. It affects males and females equally. In addition, some individuals may have mosaic NF, in which some but not all cells of the body carry the mutation.