
Longest common subsequence problem. The longest common subsequence (LCS) problem is to find the longest subsequence common to all sequences in a set of sequences (often just two).
(Note that a subsequence is different from a substring, for the terms of the former need not be consecutive terms of the original sequence.) It is a classic computer science problem, the basis of file comparison programs such as diff, and has applications in bioinformatics. Complexity[edit] For the general case of an arbitrary number of input sequences, the problem is NP-hard.[1] When the number of sequences is constant, the problem is solvable in polynomial time by dynamic programming (see Solution below).
Assume you have sequences of lengths . Hirschberg's algorithm. Damerau–Levenshtein distance. While the original motivation was to measure distance between human misspellings to improve applications such as spell checkers, Damerau–Levenshtein distance has also seen uses in biology to measure the variation between DNA.

Algorithm[edit] Adding transpositions sounds simple, but in reality there is a serious complication. Presented here are two algorithms: the first,[6] simpler one, computes what is known as the optimal string alignment[citation needed] (sometimes called the restricted edit distance[citation needed]), while the second one[7] computes the Damerau–Levenshtein distance with adjacent transpositions. Bitap algorithm. The bitap algorithm (also known as the shift-or, shift-and or Baeza-Yates–Gonnet algorithm) is an approximate string matching algorithm.
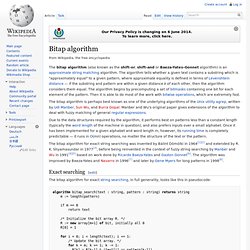
The algorithm tells whether a given text contains a substring which is "approximately equal" to a given pattern, where approximate equality is defined in terms of Levenshtein distance — if the substring and pattern are within a given distance k of each other, then the algorithm considers them equal. The algorithm begins by precomputing a set of bitmasks containing one bit for each element of the pattern. Then it is able to do most of the work with bitwise operations, which are extremely fast. Due to the data structures required by the algorithm, it performs best on patterns less than a constant length (typically the word length of the machine in question), and also prefers inputs over a small alphabet.
Exact searching[edit] The bitap algorithm for exact string searching, in full generality, looks like this in pseudocode: Fuzzy searching[edit] Needleman–Wunsch algorithm. The Needleman–Wunsch algorithm is an algorithm used in bioinformatics to align protein or nucleotide sequences.
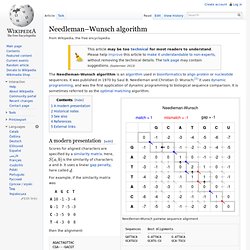
It was published in 1970 by Saul B. Needleman and Christian D. Wunsch;[1] it uses dynamic programming, and was the first application of dynamic programming to biological sequence comparison. It is sometimes referred to as the optimal matching algorithm. Needleman-Wunsch pairwise sequence alignment Sequences Best Alignments --------- ---------------------- GATTACA G-ATTACA G-ATTACA GCATGCU GCATG-CU GCA-TGCU A modern presentation[edit] Scores for aligned characters are specified by a similarity matrix. Smith–Waterman algorithm. The Smith–Waterman algorithm performs local sequence alignment; that is, for determining similar regions between two strings or nucleotide or protein sequences.
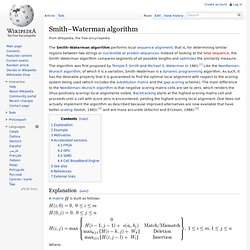
Instead of looking at the total sequence, the Smith–Waterman algorithm compares segments of all possible lengths and optimizes the similarity measure. The algorithm was first proposed by Temple F. Smith and Michael S. Matching, diffing and merging XML. Update: A newer, more complete version is here.
I've said bad things about my job working on Carleton College's website, but fundamentally it's a really sound work environment we have. Just before winter break, one of the full-time employees came to me and asked if I could make a diff between two XHTML documents for use in Carleton's CMS, Reason. This would be useful for (a) comparing versions of a document in the CMS (b) merging documents, in case two people edit the same document at the same time, so as to avoid locks and the need for manual merges.
They came to me because I told them I'd written an XML parser. I may know about XML, but I know (or rather knew) nothing about the algorithms required for such a diff and merge. A naive algorithm Here's something that won't work: a line-by-line diff and merge, along the lines of the Unix utilities diff and patch. Most obviously, this can easily break the tree structure. This XML is not well-formed! Background to a better solution Conclusion. Daisydiff - Project Hosting on Google Code. Java Notes. By Stuart D.

Gathman Last updated Apr 01, 2013 I have moved the most popular items to the top of the menu. Obsolete, but historically interesting Class Packager for Java If you want to deliver an application or applet with all the classes and resources it needs - and only the classes and resources it needs, then you need ZipLock.java. This is a utility class for examining a list of class names for all of their dependencies. When you create a JAR file with all the classes needed for your application, the JAR file is self-contained. The Object-Oriented jargon for such a self-contained application is a "Sealed System". Keep javac "hot" and ready to roll. Exploring the problems involved in comparing XML. Diffxml - XML Diff and Patch Utilities. Open Source XML Diff Written in Java.